Research at the
Integrated Materials Design Lab
Catalytic Materials

Catalysis lies at the heart of modern materials technology efforts to design efficient and sustainable energy and carbon cycles… Find out more
Electronic Materials
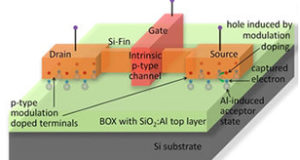
Next-generation electronics hinges on the development of new strategies for ultra large scale integration (ULSI) through sub-10nm engineering the electronic properties of semiconductors… Find out more
Energy Storage Materials

Research in this thrust will focus on a number of highly topical candidate materials for enhanced harvesting and utilization of solar energy in the context of photovoltaics… Find out more
Soft Matter Research

Recent advances in synthetic polymer chemistry have provided a wealth of complex polymer architectures such as comb polymers, star polymers and gradient copolymers… Find out more
Recent Publications
Unraveling the Factors Behind the Efficiency of Hydrogen Evolution in Endohedrally Doped C60 Structures via Ab Initio Calculations and Insights from Machine Learning Models
Understanding the origins of catalytic activity (or inactivity) in nanostructures allows for the rational design of cheap and durable catalysts. Here, consistent and comprehensive ab initio screening of endohedrally doped fullerenes as potential catalysts for hydrogen evolution reactions is performed. By examining variations in the electronic structure of the carbon atoms in the presence of the dopant, and by relying on machine learning algorithms, the origin of enhanced activity in fullerenes can be underpinned. The effect is attributed to the formation of free radicals by weakening the C─C double bonds. A number of electronic descriptors are discussed which can be fed into machine learning models to efficiently and reliably predict catalytic activities. This allows for a generalization of trends and a predictive ability that could be applied to other fullerene structures.
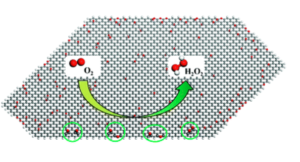
Understanding the high activity of mildly reduced graphene oxide electrocatalysts in oxygen reduction to hydrogen peroxide
The direct electrochemical synthesis of hydrogen peroxide (H2O2) would provide an attractive alternative to the traditional anthraquinone oxidation process for continuous on-site applications. Its industrial viability depends greatly on developing cost-effective catalysts with high activity and selectivity. Recent experiments have demonstrated that mildly reduced graphene oxide (mrGO) electrocatalysts exhibit highly selective and stable H2O2 formation activity [e.g., H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290]. However, the identification of active site structures for this catalytic process on mrGO is doubtful. Herein, by means of first-principles calculations, we examine the H2O2 formation activities of the active site structures proposed in experiments and find that their activities are actually very low. Then, we systematically investigate the H2O2 formation activities of different oxygen functional group structures on mrGO based on experimental observations, and discover two types of oxygen functional group structures (2EP and 1ET + 1EP) that have comparable or even lower overpotentials (<0.10 V) for H2O2 formation compared with the state-of-the-art PtHg4 electrocatalyst. Our theoretical results reveal that the graphene edge and the synergetic effects between different oxygen functional groups are essential for the superior performance of mrGO for H2O2 production. This work not only provides a feasible explanation of the cause of high H2O2 formation activity of mrGO but also offers a guide for the design, synthesis, and mechanistic investigation of advanced carbon-based electrocatalysts for effective H2O2 production.
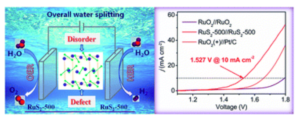
Pyrite-type ruthenium disulfide with tunable disorder and defects enables ultra-efficient overall water splitting
The exploration of efficient electrocatalysts for both the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) is significant for water splitting associated with the storage of clean and renewable energy. Here, we report a simple and scalable low-temperature sulfuration method to achieve simultaneous modulation of disorder and defects in pyrite-type RuS2 nanoparticles to dramatically enhance the HER and OER catalytic activity. The disordered structure can increase the electrochemically active surface area, while defect engineering can effectively regulate the electronic structure and thus improve the intrinsic activity, as revealed by combined experimental and theoretical density functional theory (DFT) investigations. Through controllable disorder and defect engineering, the optimized RuS2-500 catalyst (with a sulfuration temperature of 500 °C) supported on a glassy carbon electrode exhibits ultra-efficient bifunctional electrocatalytic activity with η−10 = 78 mV for the HER and η10 = 282 mV for the OER, superior to various Ru-based and pyrite-type catalysts. Remarkably, when used as both the anode and the cathode in an alkaline water electrolyzer, RuS2-500 delivers 10 mA cm−2 at an ultralow cell voltage of 1.527 V with long-term stability, outperforming the benchmark Pt/C//RuO2 couple and most state-of-the-art overall-water-splitting electrocatalysts ever reported. This work thus provides a new and facile way for improving the catalytic activity through a synergistic modulation strategy.
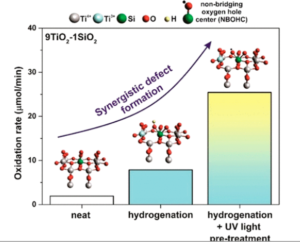
Light-Induced Synergistic Multidefect Sites on TiO2/SiO2 Composites for Catalytic Dehydrogenation
Here, we demonstrate that structural defects can induce catalytic reactivity in simple metal oxides to deliver cost-effective alternatives to noble metal group catalysts. We detail a strategy for introducing multiple defect sites in a binary TiO2–SiO2 composite to invoke synergism for oxygen activation. Hydrogenation and UV light pretreatment were applied to generate two distinct and adjacent defect sites, Ti3+ and silica-based nonbridging oxygen hole centers (NBOHC)—which work in unison to activate oxygen and oxidize formic acid under ambient conditions without light. The hydrogenation step was found to be crucial for rupturing Ti–O–Si bonds while first-principles calculations indicated that Si-doped TiO2 lowered the energy barrier for oxygen activation and formic acid dehydrogenation on the defect sites. Activity lost during the reaction was recoverable by catalyst reillumination. Defective metal oxides represent an appealing prospect in the pursuit of simple and readily accessible catalyst materials.
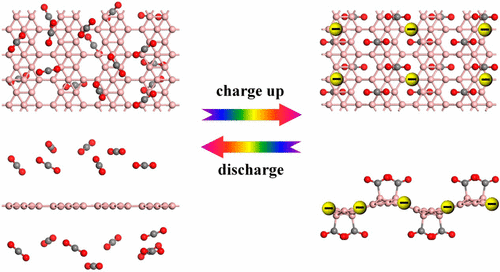
Borophene as a Promising Material for Charge-Modulated Switchable CO2 Capture
Ideal carbon dioxide (CO2) capture materials for practical applications should bind CO2 molecules neither too weakly to limit good loading kinetics nor too strongly to limit facile release. Although charge-modulated switchable CO2 capture has been proposed to be a controllable, highly selective, and reversible CO2 capture strategy, the development of a practical gas-adsorbent material remains a great challenge. In this study, by means of density functional theory (DFT) calculations, we have examined the possibility of conductive borophene nanosheets as promising sorbent materials for charge-modulated switchable CO2 capture. Our results reveal that the binding strength of CO2 molecules on negatively charged borophene can be significantly enhanced by injecting extra electrons into the adsorbent. At saturation CO2 capture coverage, the negatively charged borophene achieves CO2 capture capacities up to 6.73 × 1014 cm-2. In contrast to the other CO2 capture methods, the CO2 capture/release processes on negatively charged borophene are reversible with fast kinetics and can be easily controlled via switching on/off the charges carried by borophene nanosheets. Moreover, these negatively charged borophene nanosheets are highly selective for separating CO2 from mixtures with CH4, H2, and/or N2. This theoretical exploration will provide helpful guidance for identifying experimentally feasible, controllable, highly selective, and high-capacity CO2 capture materials with ideal thermodynamics and reversibility.
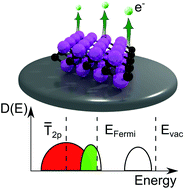
The origin of low workfunctions in OH terminated MXenes
The workfunction is an important parameter that governs several electronic phenomena occurring at the surfaces and interfaces of materials. Here, we study MXenes, which are two dimensional metal carbides and nitrides. The workfunction is strongly dependent on the terminating functional groups which induce surface dipoles and Fermi level shifts. Here, we establish a correlation between the workfunction and the adsorbate’s 2p band centres. Focusing on the OH terminated MXenes which have intrinsically low workfunctions, we show that a rigid relation between the 2p band centres and workfunctions exists which resembles a volcano plot. This imposes a limit on the lowest possible workfunctions of ∼1.2 eV and sets an optimum value of the 2p band centres at which this low workfunction can occur which we determined to be ∼−5.45 eV relative to the Fermi level. We demonstrate that neither strain modulation nor doping can achieve workfunctions lower than this.
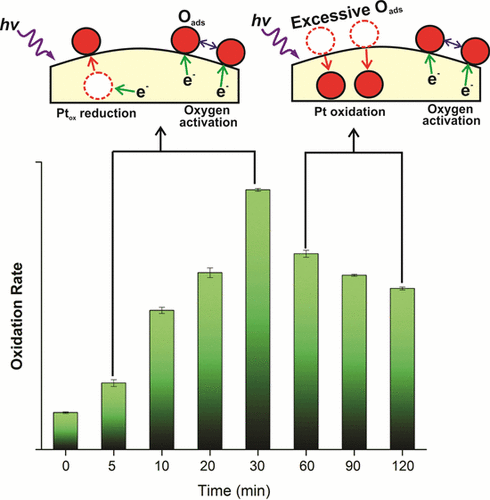
Light, Catalyst, Activation: Boosting Catalytic Oxygen Activation Using a Light Pretreatment Approach
Oxygen activation is a key reaction step in assorted thermal-catalytic processes. Here we use light pretreatment to boost oxygen activation by platinum, palladium, or gold loaded on TiO2. Light pretreatment improved catalyst performance by altering metal oxidation state and/or activated oxygen (Oads) generation. For Pt/TiO2 and Pd/TiO2, light pretreatment initially promoted Oads accumulation on the metal. In time, the metal deposits underwent oxidation with a concomitant decrease in the light pretreatment benefit. Au/TiO2 differed in that no Au oxidation was observed. Electrocatalytic assessment indicated that light pretreatment lowered the energy for oxygen activation and that Au was less able to activate oxygen overall. First-principles calculations demonstrated that light pretreatment reduces the oxygen dissociation barrier as each metal becomes negatively charged via electron injection from the TiO2 conduction bands. The calculations also illustrated the ability of Pt and Pd to stabilize the oxygen once dissociated, a trait not imbued by Au.